
Nobel Women

by Meeri Kim
Just two years after the inaugural set of Nobel Prizes were bestowed in 1901, Marie Curie snagged one-quarter of the Physics award for her work on radioactivity [1]. She became the first woman to receive the Nobel Prize and, less than a decade later, the Nobel Committee recognised her work in chemistry with a second award. Curie still holds the distinction today as the only person to be awarded the Nobel Prize in two different scientific fields [2].
In the more than one hundred years that followed, women continued to receive Nobel Prizes — but at a much lower rate than their male counterparts. Between 1901 and 2019, the world’s most prestigious awards have only been given to 53 women, as compared to a whopping 897 men [3]. The majority of female Laureates won for their contributions to Literature or Peace, while only 21 female Laureates have been recognized in the scientific fields (Physics, Chemistry, Physiology or Medicine, Economic Sciences) [4].

Clearly, women remain under-represented among Nobel Laureates, particularly in the sciences. Fortunately, some positive changes are afoot. The organizations in charge of the science prizes — the Royal Swedish Academy of Sciences and the Nobel Assembly at the Karolinska Institute in Stockholm — have started to alter their nomination processes to support gender diversity [5]. In the last 16 years, eleven women were honoured with the Nobel Prize in the scientific fields, which is one more than the entire first century of the award’s history [6]. If taken as a trend, the next 100 years should see the addition of many more female scientists to the distinguished list of Laureates.
Marie Curie: A Pioneer for Women in Science
Marie Curie overcame countless barriers against women in science to reach her level of achievement. Even though she was brilliant as a child, no university in Poland, her home country, accepted women at the time. So, she began work as a private tutor, one of the few intellectual jobs open to women. Curie also took classes at an illegal, clandestine university for women, which had scholars and scientists meet with women secretly in apartments, homes, and shops to give them an advanced education [7].
Her cousin, who directed the Museum at the Ministry of Industry and Agriculture in Warsaw, agreed to teach her wet bench chemistry — and that’s what sparked her lifelong love for experimental laboratory research. Eventually, Curie amassed enough money working as a governess to enrol at the Sorbonne. In Paris, she met husband Pierre Curie, who was a laboratory supervisor and allowed her to freely perform research in his space.
Their collaborations led to sharing half of the 1903 Nobel Prize in Physics, after studying the radioactivity of the mineral pitchblende. Her work garnered immediate attention from the scientific community, who marvelled both at the results and at the fact that a married couple made the discovery together [8]. The following clip from a Mini-Lecture describes Marie Curie’s early accomplishments that led to her first Nobel Prize.
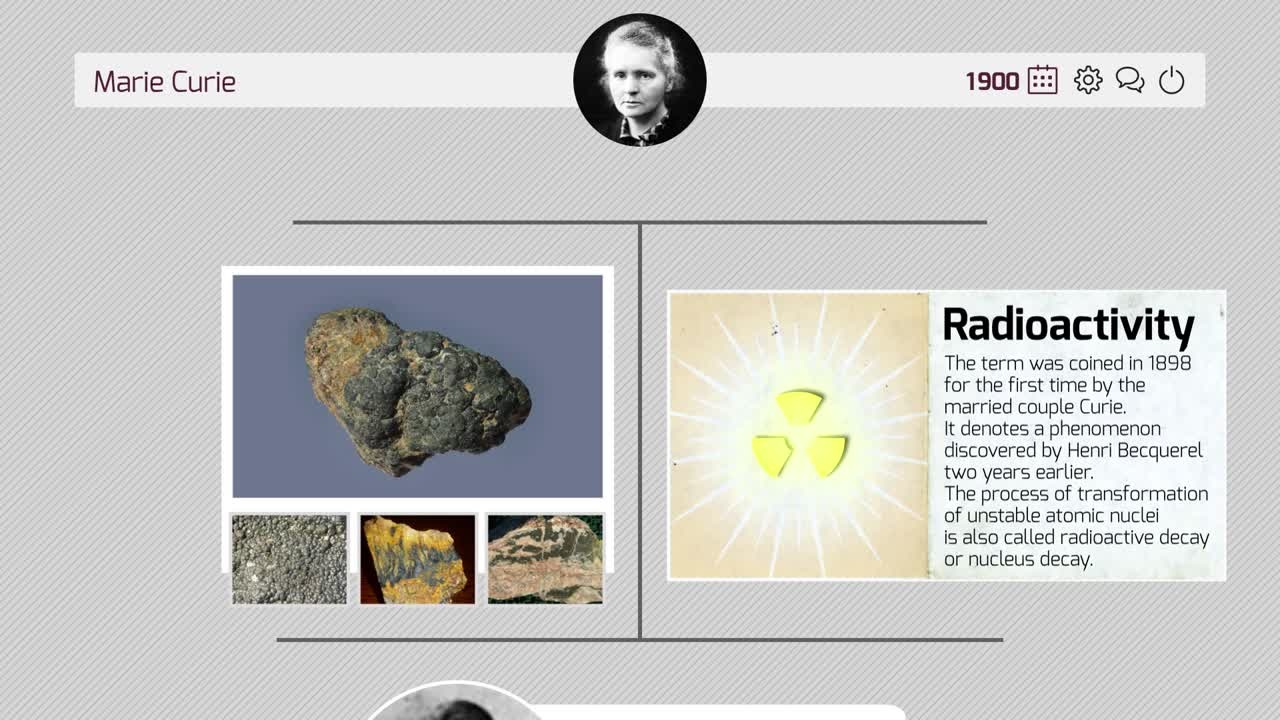
(00:03:05 - 00:04:43)
The complete video is available here.
Even after her husband’s death in 1906, Curie persisted with her research on pitchblende despite falling into a period of immense grief. She became the first woman to lecture at the Sorbonne, and in 1911, received her second Nobel Prize in Chemistry for the discovery of the radioactive elements polonium and radium contained within pitchblende. One of her two daughters, Irène Joliot-Curie, followed closely in her mother’s footsteps by working on radioactivity and even winning a Nobel Prize [10]. She shared the 1935 Nobel Prize in Chemistry with her husband, Frédéric Joliot, for the synthesis of new radioactive elements.
1940 - 1979: The Next Generation of Pioneers
After Irène Joliot-Curie’s Nobel Prize, twelve long years passed before another female scientist would be recognised by the Nobel Committee. In 1947, Gerty Cori shared the Nobel Prize in Physiology or Medicine with her husband, Carl Cori, and Bernardo Houssay [11]. They studied mechanisms of metabolism together, including how glycogen is converted into glucose. The Coris were honoured for their identification of the enzyme that initiates the decomposition of glycogen into glucose.
The 1960s finally saw two women receive Nobel Prizes for Physics and Chemistry, distinctions previously only held by the Curies. Maria Goeppert-Mayer (Physics, 1963) worked for many years in physics without pay, since she had difficulty securing a university position as a woman. Nevertheless, she stuck with her research on chemical physics and the origin of the elements. After much perseverance and help from colleagues, she nabbed one-quarter of the Nobel Prize for an atomic model in which protons and neutrons are distributed in shells with different energy levels [12].
Around the same time as Goeppert-Mayer, Dororthy Crowfood Hodgkin (Chemistry, 1964) similarly found herself unable to find work, despite graduating from Oxford University with stellar grades. Fortunately, J.D. Bernal at Cambridge took her under his wing to perform research in his X-ray crystallography lab. The technique measures the angles and intensities of diffracted X-ray beams to determine the three-dimensional structure of a crystal.
The following clip from a Mini-Lecture on X-ray crystallography describes how Bernal mentored Hodgkin and others with his idea of using the technique to analyse the structure of large biomolecules.

(00:05:29 - 00:06:53)
The complete video is available here.
Crowfoot-Hodgkin greatly advanced the technique by applying it to biomolecules like penicillin, insulin, and vitamin B12 [13-15]. She was honoured as the sole recipient of the 1964 Nobel Prize in Chemistry for her achievements. Crowfoot-Hodgkin attended the Lindau Meetings in total five times. In her final Lindau lecture, she recalls going back to Oxford after a two-year stint at Cambridge and successfully determining the structure of penicillin.

(00:55:20 - 01:01:43)
The complete video is available here.
Across the pond, a bright young woman named Rosalyn Yalow switched her major from chemistry to physics while studying at New York’s Hunter College [16]. She had recently read Marie Curie’s biography, written by her daughter Eve Curie, which spurred Yalow’s excitement about nuclear physics. Because World War II had created a shortage of male graduate students in the U.S., she was accepted into physics graduate school at the University of Illinois — the first female student in the department since 1917.
Eventually, Yalow returned to New York to perform research at the Veterans Administration Hospital in the Bronx. In collaboration with Solomon A. Berson, she pioneered a technique called radioimmunoassay, which uses radiolabelled molecules to measure small concentrations of substances in the body [17]. In particular, they added radiolabelled iodine to the insulin molecule in order to follow the hormone after injection into the body. Yalow and Berson found that diabetic patients given cattle insulin had developed an immune response and formed antibodies against it. Today, as a result of their discovery, diabetic patients only take human insulin.
She received the 1977 Nobel Prize in Physiology or Medicine, taking home half the prize (Berson had unfortunately passed away at this point). In this 1978 Lindau lecture, Yalow describes her early work on type 2 diabetes that led to the development of radioimmunoassay.

(00:02:50 - 00:06:17)
The complete video is available here.
1980 - 2008: Women Who Changed the World of Medicine
The next several decades saw a handful of female Laureates in the fields of physiology or medicine recognised for their work.
Barbara McClintock was granted an unshared Nobel Prize in 1983 for her discovery of mobile genetic elements in plants that predated the knowledge of DNA’s double helix structure [18]. She found evidence of transposable elements, or DNA sequences that can change their position within a genome, in maize [19]. However, her work was largely dismissed and ignored for thirty years, since it was assumed that genes were fixed in their position within a chromosome. When transposable elements were found in bacteria, her work was finally recognised as being correct all along.
Three years later, the Nobel Committee honoured Stanley Cohen and Rita Levi-Montalcini for their work on growth factors, or substances that regulate cell and organ growth [20]. She knew early on that a conventional life wasn’t for her, as she described in her 1988 autobiography: “My experience in childhood and adolescence of the subordinate role played by the female in a society run entirely by men, had convinced me that I was not cut out to be a wife.” [21] Her dream of attending medical school was discouraged by her father, who believed medicine was a course of study “unsuitable for a woman.”
Levi-Montalcini not only graduated summa cum laude from the Turin School of Medicine in 1936, but she also performed neurogenesis research in the lab of renowned Italian histologist Giuseppe Levi. This early work influenced her Nobel Prize-worthy discovery of nerve growth factor (NGF) in 1950, which promotes the growth and survival of mammalian nerve cells.
Her Lindau lecture in 1993, titled “The Magna Carta of Duties,” discussed how scientists have a duty to promote the survival of mankind and the preservation of the environment.

(00:00:20 - 00:05:02)
The complete video is available here.
The 1988 Nobel Prize in Physiology or Medicine focused on the innovations around drug treatment for debilitating conditions like leukaemia, gout, and hypertension [22]. Gertrude Elion shared the award with two male scientists, Sir James W. Black and her collaborator George H. Hitchings. Elion and Hitchings demonstrated differences in nucleic acid metabolism among normal cells, cancer cells, protozoa, bacteria, and viruses. Called rational drug design, they exploited these differences to develop new drugs instead of relying on trial and error. The drugs created by the team helped treat leukaemia, malaria, lupus, hepatitis, and gout.
The following clip of Elion’s 1996 Lindau talk introduces her pioneering research on azidothymidine, known as AZT, for the treatment of patients infected with the human immunodeficiency virus (HIV). AZT was the first effective treatment for HIV/AIDS, and she foreshadows how a combination therapy that includes AZT could manage the condition. One week after this lecture, the first such combination therapy — which helped turn HIV/AIDS into a chronic disease instead of a fatal one — was introduced at the International AIDS conference in Vancouver.

(00:26:35 - 00:30:07)
The complete video is available here.
Christiane Nüsslein-Volhard received the Nobel Prize in 1995, along with two male colleagues, for her work on the genetic control of early embryonic development. They studied the humble fruit fly, Drosophila melanogaster, which goes from egg to fully grown in only 9 days. Nüsslein-Volhard and Eric Wieschaus wanted to find the genes that govern this process. They randomly mutated approximately half of the egg’s genes and observed what happened to its adult body. After more than a year of gruelling experimentation, the two biologists identified 15 different genes that affected how Drosophila embryos developed [23].
She has attended five Lindau Nobel Meetings in total, with the most recent being in 2010. What follows is a clip of Nüsslein-Volhard from that meeting speaking to young scientists in a more intimate classroom forum about the genetic basis of morphological evolution. She introduces her recent research on the striped pigmentation pattern of the adult zebrafish, and how it amounts to studying "the "evolution of beauty."

(00:08:05 - 00:12:20)
The complete video is available here.
The next female Nobel Laureate was Linda Buck, who made key discoveries about the organisation of the olfactory system and received the Nobel Prize in Physiology or Medicine in 2004. The sense of smell largely remained a mystery compared to other senses at the time. So, in 1991, Buck and her colleague Richard Axel broke new ground when they published a paper demonstrating how hundreds of genes code for the odorant sensors located in the olfactory sensory neurons in our noses [24].
Four years later, the Nobel Committee honoured French virologist Françoise Barré-Sinoussi for her discovery of the retrovirus HIV in 1983. The first cases of AIDS had appeared around the world, including in France, and she was tasked with determining whether a retrovirus could be responsible for this alarming epidemic. Retroviruses carry their genetic material in the form of RNA instead of DNA, using an enzyme called reverse transcriptase to convert their RNA into DNA upon infecting a cell. Barré-Sinoussi detected significant reverse transcriptase activity in a lymph node biopsy from an AIDS patient [25].
In a video clip featured in Barré-Sinoussi’s Nobel Lab 360, she introduces the motivations of her retrovirology laboratory at the renowned Pasteur Institute, where she has performed research for more than forty years.

Explore further Nobel Labs 360° here.
She gave a Lindau lecture in 2015 on how translational research on viral infectious diseases can lead to treatment and prevention options for patients, with the past three decades of HIV/AIDS science as a highlighted example.

(00:02:03 - 00:05:42)
The complete video is available here.
2009: Four Nobel Women in a Single Year
The year 2009 proved to be a record-breaking one for women in science, as four female Nobel Laureates were added to the honoured list at once from the fields of Chemistry, Physiology or Medicine, and Economic Sciences. Most notably, Ada Yonath received the Chemistry award for her research on the structure and function of the ribosome [26]. A woman had not been awarded the Nobel Prize in Chemistry since 1964, when Dorothy Crowfoot-Hodgkin was the sole recipient for her advancements in X-ray crystallography to study the structure of biomolecules.
Similar to her predecessor Crowfoot-Hodgkin, Yonath and her colleagues employed X-ray crystallography to map the complex structure of ribosomes, which translate the information contained within DNA into the body’s thousands of proteins. These 3D models, which consist of hundreds of thousands of atoms, have applications for the development of antibiotics that target ribosome function in bacteria. Many of today’s antibiotics work in this way to treat disease.
The Israeli crystallographer has attended the Lindau Meetings nine times in total since 2010. In 2011, Yonath gave a lecture titled “Climbing the Everest Beyond the Everest,” where she described her research on ribosomes as reaching the summit of mountain only to find a taller and more difficult peak to ascend. The following clip is the closing of her talk, where she directly addressed the young women in the audience who wonder whether it’s possible to do science and have children.

(00:40:25 - 00:41:26)
The complete video is available here.
Two women, Elizabeth Blackburn and Carol Greider, shared the 2009 Nobel Prize in Physiology or Medicine with a male colleague, Jack Szostak, for the discovery of how chromosomes are protected by telomeres and the enzyme telomerase [27]. Early studies on the fruit fly and maize revealed special structures at the ends of chromosomes, called telomeres, that prevent them from fusing to one another.
In 1982, Blackburn and Szostak observed that a DNA sequence repeated several times at the end of chromosomes from Tetrahymena, a unicellular ciliate organism, had a protective effect against chromosome degradation. Later, the repeating sequence was found to be telomere DNA, which is present in most plants and animals. Research in the 1970s brought up the end-replication problem associated with telomeres, which observed that a small piece of telomeric DNA is lost after each cell division [28]. Eventually, cell death would occur after all the telomeric DNA was gone.
In reality, however, the number of repeating sequences at the ends of chromosomes varied throughout its lifetime, both shrinking and growing. In the following clip from her 2015 Lindau lecture, Blackburn describes how work with her graduate student, Greider, led to the discovery of telomerase, which extends telomere DNA.

(00:03:50 - 00:06:09)
The complete video is available here.
Lastly, one more glass ceiling shattered in 2009, as Elinor Ostrom received the Sveriges Riksbank Prize in Economic Sciences in Memory of Alfred Nobel — a first for any woman since the honour was first awarded in 1969 [29]. A decade later, French-American economist Esther Duflo would follow in Ostrom’s footsteps by receiving the 2019 prize for her experimental approach to alleviating global poverty.
2010 and Beyond: Standing on the Shoulders of Giants
The 2010s saw a continuation of 2009’s successful trend for women in science. May-Britt Moser and her husband Edvard Moser received the 2014 Nobel Prize in Physiology or Medicine, along with American-British neuroscientist John O’Keefe — one of six married couples throughout history where both partners have won the esteemed award [30]. The Mosers identified grid cells in the brain, a type of nerve cell that generates a coordinate system and serves as our “inner GPS” [31].
In a video clip featured in the Moser’s Nobel Labs 360°, May-Britt Moser describes the patterns of activity she observed in a part of the brain called the entorhinal cortex, which helps rats — and humans — find their way around a spatial environment.

Explore further Nobel Labs 360° here.
One year after the Mosers won their award, Chinese pharmaceutical chemist Tou Youyou received a half-share of the 2015 Nobel Prize in Physiology or Medicine for her work in malaria [32]. Her discovery of the drug Artemisinin has significantly reduced the mortality rates for patients around the world suffering from the mosquito-borne disease.
Interestingly, Youyou was reading through ancient Chinese literature on herbal medicine when she found clues about an active component from sweet wormwood (Artemisia annua). She discovered a low-temperature extraction process that effectively isolated an antimalarial substance and volunteered to be the first human subject. It worked, and after successful clinical trials with malaria patients, Youyou presented her results to the World Health Organization in 1981. Today, Artemisinin is used in all parts of the malaria-ridden world and saves more than 100,000 lives each year in Africa alone.
In 2018, female scientists nabbed the Nobel Prizes for both Chemistry and Physics. American chemical engineer Frances Arnold took home half of the Chemistry prize for conducting the first directed evolution of enzymes [33]. She introduced random mutations into an enzyme’s genetic code so it could operate in a completely different environment — one consisting of dimethylformamide (DMF), a powerful organic solvent -- instead of a water-based solution. Arnold introduced these mutations into bacteria that make the enzyme, subtilisin E, and selected the ones that performed the best in DMF. Subtilisin E, commonly found in detergents and food processing applications, could be used to synthesize peptides in DMF.
After three rounds of this type of artificial selection, she ended up with a variant of subtilisin E that worked 256 times better in DMF than the original enzyme. This variant had ten different mutations — a combination that would have been much more difficult or even impossible to find using brute force enzyme engineering.
The Nobel Committee hadn’t awarded the Physics prize to a woman since Maria Goeppert-Mayer in 1963. Fifty-five years later, Canadian optical physicist Donna Strickland broke the dry spell when she received the Nobel Prize for her creation of ultrashort high-intensity laser pulses with colleague Gérard Mourou [34]. They shared half of the award with American physicist Arthur Ashkin, who invented optical tweezers.
The intensity of short laser pulses had reached a wall in the mid-1980s. They could not be made any more intense without destroying the amplifying material — until Strickland and Mourou invented a novel technique called chirped pulse amplification (CPA). Instead of amplifying the short pulse straight away, CPA involved stretching it in time first, then amplifying the pulse and compressing it. This method allowed for the creation of the shortest and most intense laser pulses ever made, and CPA went on to have important applications in physics, chemistry, and medicine.
In this video clip from the 69th Lindau Nobel Laureate Meeting, Strickland shares her impressions of her first Lindau Meeting and interacting with the young scientists.

More impressions of Nobel Laureates are available here.
All of the female Nobel Laureates throughout history come from incredibly diverse backgrounds and worked in vastly different disciplines, but they share several common traits that spell success in science: creativity, persistence, passion, and vision [35]. Being one of a few — or even the only — woman in a male-dominated environment can be intimidating and isolating. But for these women, the greater, overarching goal of scientific discovery kept them on a steady track that eventually led to the most prestigious award in their fields.
The opportunities for women in science have certainly improved since the days of the first Nobel Prizes, thanks to the herculean efforts of such pioneers as Marie Curie, Irène Joliot-Curie, Maria Goeppert Mayer, and Dororthy Crowfood Hodgkin. And today, the latest generation of Laureate women — Ada Yonath, Elizabeth Blackburn, May-Britt Moser, Donna Strickland, and others — continue the tradition of giving back by inspiring the budding young female scientists around the world.
Bibliography
[1] Facts about Marie Curie (Physics) by nobelprize.org
[2] Charyton, C. et al. (2011) Gender and Science: Women Nobel Laureates. Journal of Creative Behavior. Vol. 45, 203-214
[3] Facts about the Nobel Prize in general by nobelprize.org
[4] Facts about Nobel Prizes awarded to women by nobelprize.org
[5] Gibney, E. (2018) What the Nobels are — and aren’t — doing to encourage diversity. Nature. Vol. 562, 19
[6] Lunnemann, P., Jensen, M.H. & Jauffred, L. (2019) Gender bias in Nobel Prizes. Palgrave Communications. Vol. 5, 46
[7] Rockwell, S. (2003) The Life and Legacy of Marie Curie. Yale Journal of Biology and Medicine. Vol. 76, 167-180
[8] Quinn, S. (2011) A Test of Courage: Marie Curie and the 1911 Nobel Prize. Clinical Chemistry. Vol. 57, 653-658
[9] Facts about Marie Curie (Chemistry) by nobelprize.org
[10] Facts about Irène Joliot Curie by nobelprize.org
[11] Facts about Gerty Cori by nobelprize.org
[12] Mayer, M.G. (1949) On Closed Shells in Nuclei. II. Physical Review. Vol. 75, 1969-1970
[13] Hodgkin, D.C. (1949) The X-ray analysis of the structure of penicillin. Advancement of Science. Vol. 6, 85-89
[14] Hodgkin, D.C. (1972) The Structure of Insulin. Diabetes. Vol. 21, 1131-1150
[15] Hodgkin, D.C. et al. (1957) The Structure of Vitamin B12 I. An Outline of the Crystallographic Investigation of Vitamin B12. Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences. Vol. 242, 228-263
[16] Rosalyn Yalow - sciencehistrory.org
[17] Yalow, R.S. and Berson, S.A. (1960) Immunoassay of endogenous plasma insulin in man. The Journal of Clinical Investigation. Vol. 39, 1157-1175
[18] Press release for the 1983 Nobel Prize in Physiology or Medicine
[19] McClintock, B. (1953) Induction of instability at selected loci in maize. Genetics. Vol. 38, 579-599
[20] Press release for the 1986 Nobel Prize in Physiology or Medicine
[21] Rita Levi Montalcini at jax.org
[22] Press release for the 1988 Nobel Prize in Physiology or Medicine
[23] Nüsslein-Volhard, C., Wieschaus, E., and Kluding, H. (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Wilhelm Roux's archives of developmental biology. Vol. 193, 267–282
[24] Buck, L. and Axel, R. (1991) A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell. Vol. 65, 175-187
[25] Biography of Francoise Barré-Sinoussi at nobelprize.org
[26] Schluenzen, F. et al. (2000) Structure of Functionally Activated Small Ribosomal Subunit at 3.3 Å Resolution. Cell. Vol. 102, 615-623
[27] Press release for the 2009 Nobel Prize in Physiology or Medicine
[28] Varela, E. and Blasco, M.A. (2010) 2009 Nobel Prize in Physiology or Medicine: telomeres and telomerase. Oncogene. Vol. 29, 1561-1565
[29] Summary of the Sveriges Riksbank Prize in Economic Sciences in Memory of Alfred Nobel 2009 by nobelprize.org
[30] Nobel Prize awarded couples by nobelprize.org
[31] Hafting, T., Fyhn, M., Molden, S., Moser M.-B., and Moser, E.I. (2005) Microstructure of a spatial map in the entorhinal cortex. Nature. Vol. 436, 801-806
[32] Biography of Tu Youyou by nobelprize.org
[33] A Revolution in Chemistry by nobelprize.org
[34] Summary of the 2018 Nobel Prize in Physics
[35] Women who changed the world by nobelprize.org