
Model Organisms

by Neysan Donnelly
As the term suggests, model organisms are used to study processes, diseases and phenomena that cannot be easily examined in other, usually more complex, organisms. The logic behind the use of model organisms rests on the acceptance that all life on earth has evolved from one single source and shares one single ancestor. This means that genetic, cellular and biochemical mechanisms are shared across living beings, from bacteria to humans. Many important molecules and processes evolved relatively early in history and were then “expanded on” and refined in more complex organisms. Thus, for example, insights gleaned by studying molecules and processes in very simple organisms can have important implications for human disease.
While the use of model organisms has really taken off in the last 100 years, important scientific discoveries were made using model organisms already in the 19th century. Perhaps the most striking example was Mendel’s use of peas to elucidate the principles behind the genetics of heredity.
One of the most commonly employed model organisms is the humble baker’s yeast, the unicellular Saccharomyces cerevisiae. Although humans and yeast are separated by millions of years of evolutionary history, as eukaryotes, organisms that encase their nuclei within membranes, we still share many genes and mechanisms. The much simpler biology of S. cerevisiae as well as the ease with which yeast can be manipulated and altered means that the fundamental characteristics of many cellular processes can more easily studied in this organism than in humans or more complex multicellular organisms. Günter Blobel won the Nobel Prize in Physiology or Medicine in 1999 for discoveries related to intracellular protein traffic. In his lecture at the 53rd Lindau Nobel Laureate Meeting in 2003, he talks about his studies on the nuclear pore complex, a massive protein conglomerate consisting of hundreds of individual proteins that acts as the gate regulating the transport of substances in and out of the nucleus. In the following excerpt, Blobel explains why it makes sense to study such massive and complex structures in baker’s yeast.

(00:08:31 - 00:10:10)
A huge amount of Nobel Prize-winning research was performed in model organisms. In fact, according to the reckoning of the Foundation for Biomedical Research, 96 of the 108 recipients of the Nobel Prize in Physiology or Medicine used model organisms in their research (https://fbresearch.org/medical-advances/nobel-prizes/). In the context of Nobel Prize-winning research, some of these organisms have been employed only by one or a very few laureates due to their very specific characteristics, while others such as Saccharomyces cerevisiae, the nematode Caenorhabditis elegans as well as mice, have yielded many Nobel Prize-winning insights.
An example of the former is Tetrahymena thermophila, a free-living, unicellular organism commonly found in freshwater ponds. T. thermophila possesses many attributes that make it an excellent model organism. For example, its large numbers of cilia and complex cytoskeleton make it very suitable for studying these structures. Nobel Laureate Elizabeth Blackburn was drawn to the organism for other reasons, however: namely, because it contains large numbers of chromosomes and because it can be easily grown in large quantities to provide huge amounts of biological material. In the following excerpts from her lecture at the 61st Lindau Meeting in 2011, she talks about how she employed Tetrahymena to study telomeres, then collaborated with fellow future laureate Jack Szostak to study telomeres in another model organism, baker’s yeast, and subsequently used Tetrahymena again to demonstrate that the telomerase enzyme is essential.

(00:09:04 - 00:11:16)
(00:19:28 - 00:22:11)
The nematode Caenorhabditis elegans is a soil-living worm of about 1 mm in length and has played pivotal roles in the discovery and elucidation of two fundamental cellular processes: programmed cell death and RNA interference (RNAi). These organisms have a very simple anatomy (only about 1000 cells) and are even transparent; yet, they still share many important cellular processes with humans. Further, they can be easily grown in large quantities, stored in the freezer and are amenable to genetic modification. They also have very short lifespans, which means that they are excellent models in which to study lifespan within a feasible timeframe. In his lecture at the Lindau Nobel Laureate Meeting in 2007, Craig Mello, who shared the 2006 Nobel Prize with Andrew Fire for discovering RNAi, discusses the usefulness of the worm as a model organism, offers a somewhat cosmic explanation for how RNAi can be linked with the Big Bang (!) and underlines the importance of model organisms in general.

(00:09:01 - 00:15:57)
Later in his lecture, he talks more specifically about how C. elegans was used to study RNAi and to discover genes encoding the RNAi machinery:
(00:26:19 - 00:27:37)
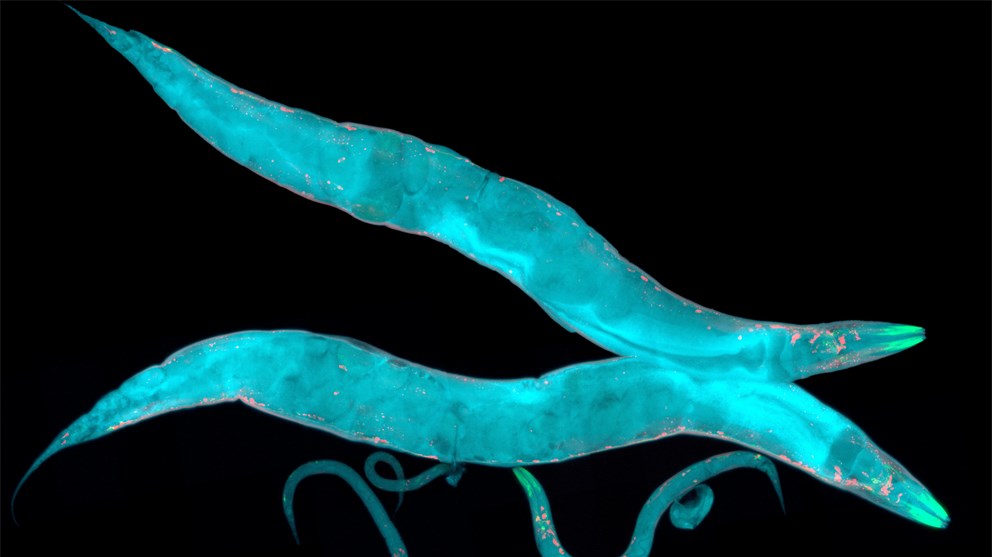
Picture/Credit: HeitiPaves/istockphoto.com
C. elegans was also instrumental in elucidating the details behind the genetic programme that sets in motion programmed cell death. In his lecture at the 60th Lindau Nobel Laureate Meeting in 2010, Robert Horvitz, who shared the 2002 Nobel Prize in Physiology or Medicine with Sydney Brenner and John Sulston, introduces C. elegans as a model organism, describes its characteristics and outlines how it was used to unravel the blueprint governing programmed cell death.

(00:08:59 - 00:12:08)
Later in his talk, he describes the eureka moment when it became clear that the genes and processes that they were studying were not only important in worms but were also significant for human biology.
(00:18:45 - 00:20:43)
The African clawed frog Xenopus laevis boasts many characteristics that make it an excellent model organism. One important advantage is that, as a vertebrate, X. laevis is much more similar to humans than many other commonly used model organisms. Also, manipulating these organisms is relatively straightforward, and this, together with the fact that female frogs can be easily induced to lay eggs means that X. laevis are excellent models in which to study development. Further facilitating research using this organism is the fact that as of 2016, the genome of X. laevis has been fully sequenced.
Peter Agre discovered the aquaporin water channels, work for which he shared the 2003 Nobel Prize in Chemistry. The critical breakthrough was made in experiments performed in frog eggs, as Agre describes in his 2011 lecture in Lindau.

(00:06:25 - 00:07:53)
Christiane Nüsslein-Volhard shared the 1995 Nobel Prize in Physiology or Medicine for discoveries related to the genetic programme behind embryonic development. This work was carried out in the fruit fly Drosophila melanogaster, an organism which is particularly amenable to genetic studies and resulted in the identification of many genes involved in building the basic body plans of embryos. However, while the early stages of embryonic development exhibit remarkable similarities even among very distantly related species, subsequent development is of course vastly different. Thus, for her studies on the next steps in organismal development, Nüsslein-Volhard turned to another model organism: the zebrafish Danio rerio.

Picture/Credit: Mirko_Rosenau/istockphoto.com
In common with X. laevis, the zebrafish is also a vertebrate, and, in fact, 70% of human genes are found in this organism. This means that many physiological processes can be well modelled in zebrafish. Further, the organisms are also easy to maintain and modify. At her lecture at the 60th Lindau Meeting, Nüsslein-Volhard described some of the other advantages of using zebrafish as a model organism and talks about the research questions she addresses studying these organisms.

(00:19:28 - 00:21:05)

Picture/Credit: Antagain/istockphoto.com
One of the very first model organisms introduced into the laboratory was the fruit fly Drosophila melanogaster, and this was largely due to the efforts of one man: Nobel Laureate Thomas Hunt Morgan. In fact, his work in fruit flies can be said to have ushered in the modern science of genetics by showing that genes are encoded on chromosomes. D. melanogaster are now one of the most common model organisms in laboratories the world over and are used to study everything from how DNA is packaged to neurodegenerative conditions. Insects are also excellent model organisms in which to study infection and immunity. In his lecture at the 64th Lindau Nobel Laureate Meeting in 2014, Jules Hoffman, who shared one half of the Nobel Prize in Physiology or Medicine in 2011 for discovering the function of Toll receptors in innate immunity, talks about how and why he used Drosophila in his research and what questions he has aimed to answer using these organisms.

(00:00:28 - 00:02:45)
He goes on to describe the landmark work in Drosophila that implicated Toll as playing a critical role in antifungal responses and how this paved the way for the discovery and characterisation of Toll receptors in mammals.
(00:10:10 - 00:12:59)
The mouse is broadly applied as a model in many areas of biological research and boasts many advantages, which include physiological similarity to humans, comparative ease of handling and the existence of a large number of inbred strains. Bruce Beutler performed his Nobel Prize-winning work in these organisms and by particularly focusing on mice found to be resistant to lipopolysaccharides, large, highly toxic molecules composed of both lipid and sugar, he discovered one of the Toll receptors in mammals.

(00:02:17 - 00:03:57)
Beutler goes on to talk about current approaches in his laboratory using mice as model organisms, and in particular describes the power of forward genetics, i.e., randomly inducing mutations and then identifying the genes responsible and strategies to examine gut homeostasis in the mouse following injury to the epithelium.
(00:06:26 - 00:10:07)
Laureate Martin J. Evans, whose Nobel Prize was awarded for work in establishing genetically modified mice as model organisms, points out how large and lucrative the market for mice models is:

(00:03:06 - 00:03:50)
Man’s best friend, the dog, also holds many advantages as a model organism, not least because they contract many of the same diseases as humans. Harald zur Hausen received his Nobel Prize in Physiology or Medicine in 2008 for his seminal work on cancer-causing viral infections, and in particular, papilloma viruses. In his lecture at the Lindau Nobel Laureate Meetings in 2011, zur Hausen talks about how outbreaks of papilloma in dog kennels in the US led him and his colleagues to develop the first successful vaccination against these viruses.

(00:11:31 - 00:12:37)
The advantage of using non-human primates as models of human disease is obvious: their high similarity to humans means that any findings made in such species are also very likely to be relevant for humans. Of course, this high degree of similarity also means that questions of ethics assume critical importance. One way in which this issue can be side-stepped is by using natural models. For example, certain species of primates naturally exhibit very high rates of infection with SIV, a virus which is very similar to HIV and which can cause an AIDS-like disease. However, due to adaptation to the virus, these primates do not generally develop disease. Françoise Barré-Sinoussi received the Nobel Prize in Physiology or Medicine in 2008 for the discovery of HIV. In this excerpt from her lecture at the Lindau Meeting in 2010, she underlines how useful such primates are for research into fighting AIDS.

(00:27:43 - 00:28:11)
Even better than non-human primates, of course, are humans themselves. If we are interested in human disease and human biology, would it not better to simply to study these processes in human subjects? For instance, even though mice are widely used to study cancer, findings can only be extrapolated to a certain extent. Mice remain significantly different from us in terms of the environments in which they have evolved and in many aspects of physiology.
One way in which human material can be used as a model is to use human cell lines. These are cells that have been isolated from human tissues and have generally been immortalised; i.e., they can continue to divide indefinitely. Even though the use of such cell lines to study the complex pathogenesis of human disease is limited, they do represent excellent models in which to probe cell biology and biochemistry. One of the most commonly used immortalised human cell line is called HeLa after Henrietta Lacks, the woman from whom the cells were isolated in 1951. Lacks died of cervical cancer and the cells were derived and expanded from her tumour without her knowledge or consent or that of her family’s. HeLa cells have been directly involved in many ground-breaking discoveries, such as the discovery of the polio vaccine and the development of the human papilloma virus vaccine by Harald zur Hausen. Why exactly did zur Hausen use Hela cells? It turns out these cells have in fact been immortalised by a virus called HPV-18.

(00:01:34 - 00:02:01)
Organoids are miniature, 3-D replicas of organs that are derived from stem cells and represent an important extension to using cell lines as a model for research. As they are grown in dishes in the lab, organoids are characterised by a high degree of tractability. On the other, they recapitulate much more closely the situation within a living organism than classical cell culture. Organoids are enabling important discoveries in fields such as tissue development, metabolism and cancer.
The profound ethical considerations are the most significant barrier to using humans themselves for experimental research. Humans cannot simply be infected with a virus for the sake of research or bred in large numbers with the aim of achieving statistically significant results. In the face of this dilemma, future Nobel Laureate Barry Marshal took matters into his own hands.

(00:21:00 - 00:24:02)
To say that modern medicine owes a large debt to model organisms would be an understatement. In fact, research as we know it would not even exist and many Nobel Prize-winning discoveries would not have been possible. They are truly the stars behind the stars. The organisms themselves are a diverse bunch, ranging from the yeast that ensures our bread rises, to soil-living worms, to flies, which is testament both to the huge breadth of questions that model organisms have been used to answer as well as to the ingeniousness of the men and women who first established these models.
So much for the past. What about the future? Huge sequencing efforts in human cells are allowing researchers to discover hitherto unknown genetic roots of disease, and the development of revolutionary techniques such as CRISPR means that it is now easier than ever to carry out experiments in human cells and potentially in humans themselves. Does this mean that the importance of model organisms will decline? To some extent, such developments may mean that more researchers will reach straight for human cell lines rather than starting in yeast, for example. However, forecasting the demise of model organisms may be premature. Indeed, new model organisms are still being established and described today. Being able to model a phenomenon or condition in a simple whole organism will likely still be a powerful tool for understanding human disease for some time to come.
Some Further Reading on Model Organisms:
Textbooks:
“Emerging Model Organisms: A Laboratory Manual, Volume 1” (2009) Cold Spring Harbor (New York): Cold Spring Harbor Laboratory Press. ISBN: 978-087969872-0
David A. Crotty and Alexander Gann (eds.), “Emerging Model Organisms: A Laboratory Manual, Volume 2” (2010) Cold Spring Harbor (New York): Cold Spring Harbor Laboratory Press. ISBN: 978-0-87969-826-3 (hc); 978-0-87969-872-0 (pb)
Journal article:
Bob Goldstein and Nicole King, “The Future of Cell Biology: Emerging Model Organisms” (2016) Trends Cell Biol. Nov; 26(11): 818–824.
Popular Science:
Jim Endersby, “A Guinea Pig's History of Biology” (2009) Harvard University Press, ISBN: 9780674032279
Rebecca Skloot, “The Immortal Life of Henrietta Lacks” (2010) Crown, ISBN 10: 0307888444, ISBN 13: 9780307888440